医療機器のヒューマンファクターとユーザビリティテスト
企業がNAMSAを選ぶ理由
カスタム研究プロジェクトで支援を受ける医療技術企業
医療機器ユーザビリティプロジェクト完了
医療機器の臨床および規制の専門家が在籍
グローバルデータベースのHCPおよび管理者参加者のパネル
医療機器開発におけるヒューマンファクターとユーザビリティテストの重要性
医療機器の開発において、ヒューマンファクターは極めて重要な役割を果たし、製品の機能性だけでなく、直感的でユーザーフレンドリーであることを保証します。設計初期段階から市販後の改良に至るまで、ヒューマンファクターを組み込むことで、メーカーはエンドユーザーのニーズを満たし、安全性を高め、全体的な有効性を向上させる機器を開発できます。このユーザー中心のアプローチは、以下のことに役立ちます。
- 製品コンセプト開発の最適化
- 潜在的なユーザビリティの問題を特定して解決する
- 規制遵守と承認
- 革新的で信頼性の高い製品を提供する
この研究は、デバイス設計、ユーザートレーニング、臨床プロトコルの改善に直接貢献します。テストが正確に実施されれば、メーカーはユーザビリティの問題の 97% を特定できます。
NAMSA では、ヒューマンファクターの重要性を理解しており、真に効果のある医療機器の設計とテストの支援に全力で取り組んでいます。
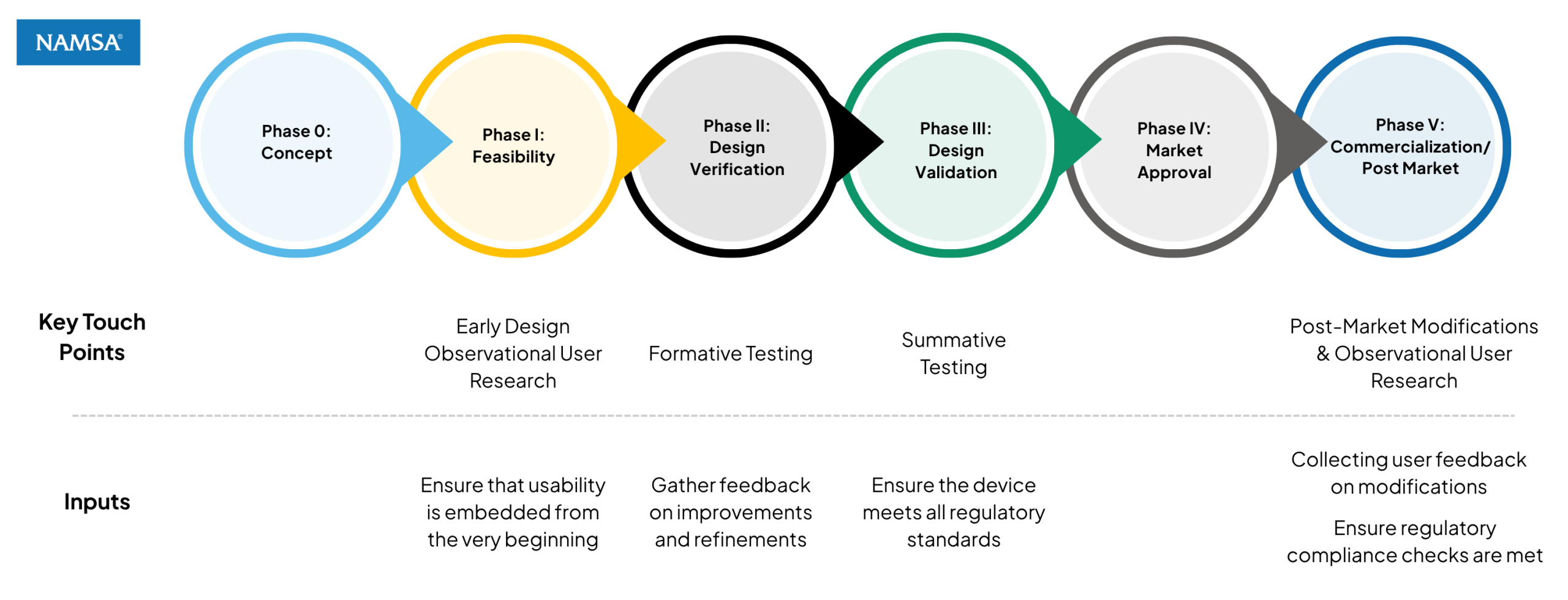
ヒューマンファクターとユーザビリティテストにおける重要なタッチポイント
包括的なユーザビリティと規制遵守を確保するため、製品ライフサイクルの様々な段階でヒューマンファクターを組み込むことができます。 NAMSA ヒューマンファクターテストをサポートできます:
- 初期設計段階: 開発の初期段階において、当社のヒューマンファクター専門家は、デバイスエンジニアやヒューマンファクターデザイナーと緊密に連携します。この段階では、初期の製品コンセプト、ワイヤーフレーム、デザインスケッチを作成し、ユーザビリティが最初から組み込まれていることを確認します。
- プロトタイプ開発: 次に、エンジニアと潜在的ユーザーを対象に、形成的テストを実施します。この段階では、プロトタイプ、モックアップデバイス、仮想フォーマットに関する貴重なユーザーフィードバックを収集し、反復的な改善と改良を行います。
- 市場承認: 製品の完成が近づくと、最終的なエンドユーザーグループを対象に総括テストが実施されます。この厳格なフェーズには、正式なユーザビリティテストプロトコル、多角度ビデオ録画、そしてデバイスがすべての規制基準を満たしていることを確認するための詳細な観察が含まれます。
- 規制基準:
- 医療機器へのヒューマンファクターとユーザビリティエンジニアリングの適用:FDAガイダンス(2016年)
- IEC 62366-1: 医療機器のユーザビリティエンジニアリング
- IEC 14971: 医療機器へのリスク管理の適用
- 市販後の変更: 製品発売後も、ヒューマンファクターは依然として重要です。デバイスのアップグレードや機能変更に関するテスト、変更に関するユーザーフィードバックの収集、ユーザビリティテストの結果、そして規制遵守チェックの遵守状況の確認などを実施しています。
Take the Next Step
ヒューマンファクターとユーザビリティに関する当社の経験について詳しく知りたいですか?
ユーザビリティテストに関する規制要件
医療機器が想定されるユーザー、用途、そして環境において安全かつ効果的であることを保証するには、ヒューマンファクターとユーザビリティテストが不可欠です。米国食品医薬品局(FDA)は、特にユーザーエラーが重大な危害につながる可能性のある高リスク機器について、これらの試験を義務付けています。FDAのガイドラインでは、市販前承認プロセスの一環としてヒューマンファクター検証試験の重要性が強調されており、新製品の市場投入を目指すメーカーにとって、ユーザビリティテストは重要なステップとなっています。
医療機器メーカーは、FDAの要件に加え、欧州連合(EU)の医療機器規則(MDR)や体外診断用医薬品規則(IVDR)、オーストラリアの医薬品行政局(TGA)といった他の地域規制にも準拠する必要があります。これらの規制も同様に、機器の安全性と有効性を確保するためのユーザビリティテストの重要性を強調しており、製品ライフサイクル全体にわたる包括的なヒューマンファクター研究を義務付けています。
NAMSAは国際規格に準拠し、ヒューマンファクターに関する世界的な規制要件の詳細とニュアンスに精通しているため、あらゆる規制へのコンプライアンスをサポートします。当社のチームは、お客様のデバイスが必要なガイドラインをすべて満たしていることを保証し、スムーズな市場参入を促進し、最高水準の安全性と使いやすさを確保します。
信頼 NAMSA ヒューマンファクターテストを実行する
NAMSAは、医療機器向けの卓越したヒューマンファクターおよびユーザビリティテストサービスの提供に誇りを持っています。私たちの独自の価値提案は、いくつかの重要な強みに基づいています。
- お客様の特定のニーズに合わせて、創造的で費用対効果が高く、効率的なテストソリューションを提供する当社の能力
- 医療専門家と患者の広大なネットワークにより、迅速かつ確実な参加者募集を実現しています。
- 当社の深い規制知識により、お客様の製品が必要なコンプライアンス基準をすべて満たしていることが保証され、スムーズな市場参入が促進されます。
- 当社の協力的なアプローチと行動調査スキルにより、洞察力のある分析と実用的な推奨事項を提供し、デバイスがコンプライアンスに準拠しているだけでなく、直感的で使いやすいことを保証します。
NAMSAをお選びいただくことで、医療機器の安全性、有効性、そしてユーザーエクスペリエンスの向上に尽力するパートナーを得ることができます。創造性、規制に関する専門知識、そして協力的なサポートへのコミットメントにより、お客様の製品は規制遵守だけでなく、直感的で使いやすいものとなり、エンドユーザーの生活に真の変化をもたらします。
よくある質問(FAQ)
FDA は当社のデバイスに対してどのようなレベルのヒューマンファクターテストを期待していますか?
FDA の期待は、デバイスのリスク プロファイル、対象ユーザー、使用環境によって異なります。 NAMSA 通常、使用関連リスク分析、デバイス分類、および過去のHFに関する調査を審査し、形成的研究、完全なバリデーション研究、または追加の正当化が必要かどうかを判断します。また、デバイスがFDAの「クリティカルタスク」フレームワークに該当するかどうかも評価します。このフレームワークは、試験の深度を決定します。その目的は、HFプログラムがFDAのガイダンスに準拠していることを保証し、審査中に予期せぬ事態を回避することです。
どのように NAMSA 適切なユーザー グループ、環境、重要なタスクを決定しますか?
NAMSA デバイスの想定ユーザー、使用目的、リスクプロファイルを分析し、代表的なユーザーグループと現実的な使用環境を定義します。リスク分析、ラベリング、ワークフローをレビューし、誤って実行された場合に危害につながる可能性のあるタスクを特定します。これらのタスクは、バリデーションスタディで評価する必要がある「重要なタスク」となります。 NAMSA また、研究が実際の使用状況を反映し、多様性と現実性に関する FDA の期待を満たすことを保証するために、識字能力の低いユーザーやストレスの多い環境などのエッジケースも考慮します。
どのようなサンプルサイズと採用戦略が FDA の要件を満たすのでしょうか?
サンプル サイズは、統計的検出力ではなく、各ユーザー グループの意味のある代表性に対する FDA の期待によって決定されます。 NAMSA 検証試験では、デバイスの複雑さやリスクに応じて、通常、ユーザーグループごとに最低15名(+パイロット1名)の参加者を推奨しています。参加者の募集は、経験、人口統計、トレーニングレベルにおいて、対象ユーザーと真に一致する参加者を見つけることに重点を置いています。 NAMSA また、実使用において事前のトレーニングが必要な場合を除き、参加者が機器を熟知していることも保証します。このアプローチは、FDAが重視する現実性とユーザーの代表性を満たすのに役立ちます。
どのように NAMSA FDA 提出基準を満たすようにヒューマンファクターのドキュメントを構成するには?
NAMSA FDAが推奨する構成(デバイスの説明、ユーザープロファイル、使用環境、リスク分析、形成的研究の要約、バリデーション研究のプロトコル、結果、結論)に従ったヒューマンファクターバリデーションレポートを作成します。これらのレポートは、リスクに基づく意思決定と研究デザインを明確に結び付け、残存する使用関連リスクが許容可能であることを実証するものです。レポートには、詳細なタスク分析、参加者の人口統計情報、観察された使用エラーも含まれます。目標は、デバイスが安全かつ効果的に使用できることを示す、一貫性があり、エビデンスに基づいたストーリーを提示することです。
NAMSA によるヒューマンファクターのテストとレポートの予想されるスケジュールとコストはどれくらいですか?
タイムラインは、デバイスの複雑さ、被験者募集の難易度、試験回数によって異なります。典型的な形成的試験は6~10週間かかりますが、プロトコルの作成、必要に応じてIRB審査、被験者募集、試験、報告を含む完全な検証試験は、12~20週間かかることがよくあります。費用は、被験者数、試験場所、必要な文書のレベルによって異なります。 NAMSA マイルストーン、依存関係、予算範囲を概説した詳細なプロジェクト計画を提供するため、HF アクティビティを規制申請戦略と一致させることができます。





興味があるかもしれない関連サービス

米国FDAおよびCMSコンサルティング

EU MDR & IVDRコンサルティング